

Мягкая форма мукополисахаридоза I типа (синдром Гурлер-Шейе)
Возраст появления симптомов, скорость их усугубления, а в некоторых случаях и тип генетической мутации определяют форму мукополисахаридоза по степени тяжести: тяжелая форма (по-другому называется «синдром Гурлер») и мягкая форма (самую мягкую форму называют «синдром Шейе» и промежуточную форму с более выраженными симптомами - «Гурлер-Шейе»).1
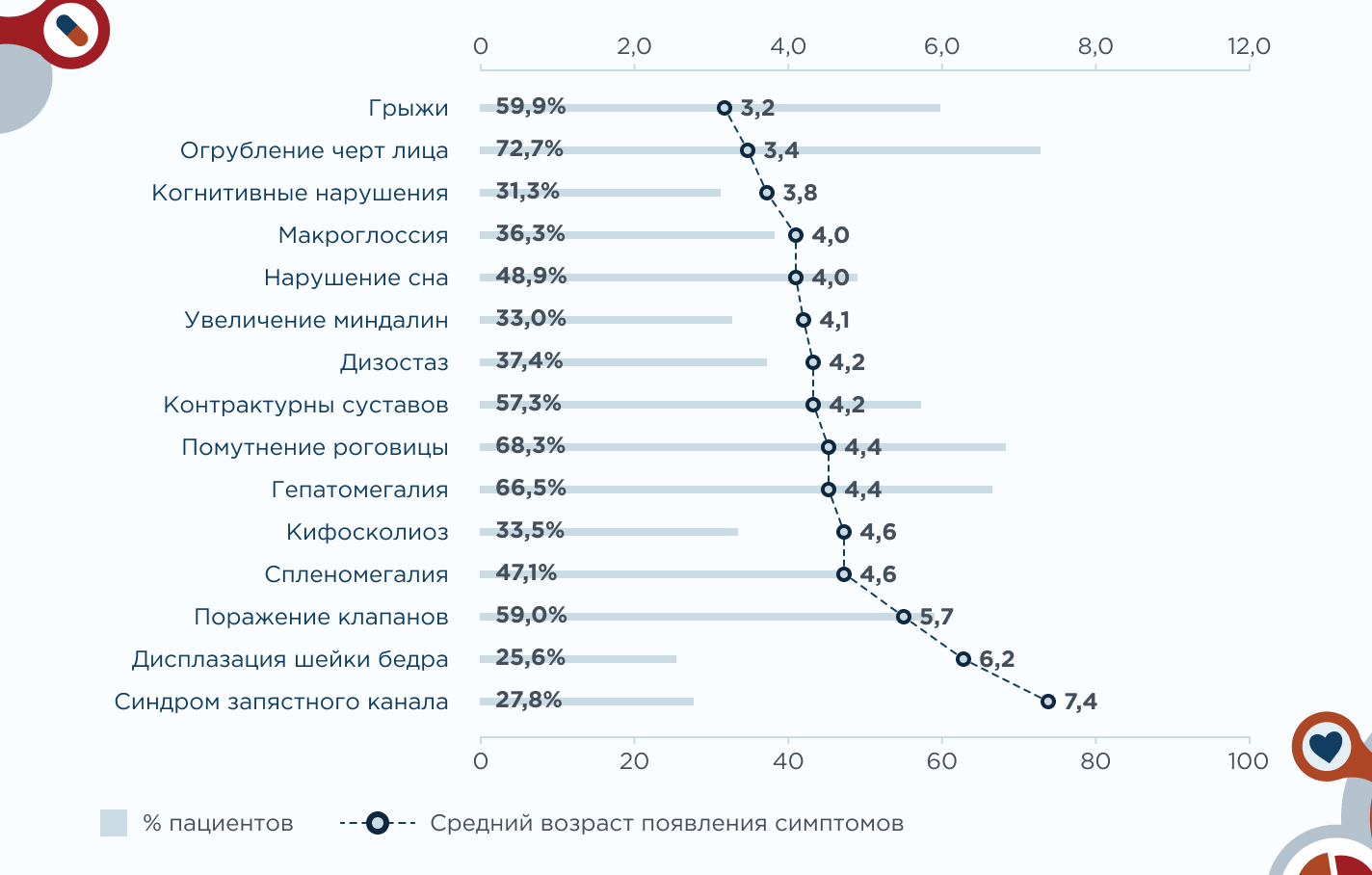
Первые проявления заболевания при синдроме Гурлер-Шейе наблюдаются в среднем в возрасте 4 лет, но могут и раньше.5
При промежуточной форме МПС I, как и при тяжелой форме, поражаются многие органы, но при этом прогрессирование заболевание происходит медленнее. Характерно умеренное отставание в физическом и интеллектуальном развитии.2
Самые частые симптомы при синдроме Гурлер-Шейе: грубые черты лица, помутнение роговицы, увеличение печени и селезенки, ограничение подвижности суставов и поражения клапанов. Характерна задержка роста.4,5
- грыжи (пупочные или паховые) встречаются у 60% пациентов с синдромом Гурлер-Шейе;4
- поражение сердца наблюдается у большинства больных (59% пациентов с синдромом Гурлер-Шейе);4
- тугоподвижность суставов наблюдается примерно у 57% пациентов с промежуточной формой и может проявляться развитием «когтистой лапы» (согнутые пальцы рук).4
У пациента с мукополисахаридозом I типа могут наблюдаться не все указанные проявления заболевания, а сочетание нескольких признаков (например, низкий рост и тугоподвижность суставов при увеличенной печени).2,4
Узнать больше: Методы диагностики и Программа лекарственного обеспечения.
Центры экспертизы

- 1. Neufeld EF & Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. 2001. McGraw Hill, New York, USA.
- 2. Michael Beck et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genetics in medicine | Volume 16 | Number 10 | October 2014 2. Stefano Bruni, The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep. 2016 Sep; 8: 67–73.
- 3. П.В. Новиков. Лизосомные болезни накопления — актуальная проблема педиатрии и современные возможности патогенетического лечения. РОССИЙСКИЙ ВЕСТНИК ПЕРИНАТОЛОГИИ И ПЕДИАТРИИ.
- 4. S.Bruni et al. The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep. 2016 Sep; 8: 67–73.
- 5. Клинические рекомендации: Мукополисахаридоз I типа у детей, 2016. Утверждены Союзом Педиатров России. Доступ: pediatr-russia.ru от 26.02.2019.